
OCUL(L) - SOL-1
I hope we see this binary correctly


Co-Author: Adu Subramanian Substack X
TOC
Intro
Pathology
Market History
TKI overview
History: Graybug
History: Clearside
Preclin
SAB-1 vs Form IV
Axpaxli clinical data
SOL-1
Inclusion criteria notes/KOD/Vabysmo explanation
Control group analysis
Axpaxli Group analysis
Sol-1 Bar
Safety
Market Possibility
Market headwinds
General Risks
Conclusion
Thesis:
Management has guided to a 70% rescue free rate vs a 20% rescue free rate, Tx vs control. I think the numbers come in closer to 90% vs 50%. The outperformance extends to 12 months and they can file on 1 trial for a superiority label. Furthermore, I suspect the EYPT data won’t be as good and thus OCUL has a better program. If successful, the arc of wAMD towards less injections bends the 15B+ market to OCUL. Furthermore, I view an outright failure as extremely unlikely (though not impossible) thus am willing to risk a “binary”.
**Intro
The Wet Age Related Macular Degeneration market is full of great therapies which have preserved and improved vision for millions since the early 2000s. Despite multiple highly efficacious drugs on the market, the discontinuation rate within the first year is 40%. These extremely safe drugs are proven to immediately improve vision in the short term and delay blindness in the long term and almost half of patients still quit early on.
Why?
All of these drugs are administered by an uncomfortable eye injection at a scheduled visit to an ophthalmologist. In many areas across America it is difficult to get an appointment and even harder to reschedule. If your treatment regimen is an Eylea injection 6 times a year, you will schedule an appointment 4 months in advance. Depending on the volume in a practice, a patient may have to wait months for a rescheduled injection, and give up altogether. The current therapies force people to schedule their lives around a hated injection, so many patients just quit.
Axpaxli is an implant in development by Ocular Therapeutix which sits in the vitreous and secretes the TKI axitinib over ~9 months. Ocular is about to readout a pivotal phase 3 trial measuring how long Axpaxli prevents patients from needing a supplemental anti-VEGF injection. Axpaxli has the possibility of being approved for every 6-12 month dosing and decreasing the dosing burden for a large basket of patients. Based on trial design and Axpaxli’s preclinical data, it should remain efficacious until the primary endpoint at 9 months and convey impressive efficacy.
**Pathology/ why VEGF Works
Wet Age Related Macular Degeneration (wAMD) is the growth of new blood vessels in the choroid of the eye. These blood vessels, formally known as the CNV(choroidal neovascularization) grow through the Bruch’s membrane and eventually the RPE causing structural damage to the eye. The neovascularization originally occurs when someone(normally of an older age) accumulates too many trash particles in the Bruch’s Membrane. This trash leads to hypoxia, which leads to the downstream release of VEGF. The influx of VEGF promotes angiogenesis, creating the CNV. The CNV growth leads to even more hypoxia causing a runaway effect of excess VEGF production.
VEGF-A is the key target for Wet AMD. Since it has been proven to be the main promoter of growth, all efficacious therapies in wet AMD target it in some way. The main isoform in the eye is VEGF 165. This isoform is so important because it is both small enough to diffuse into nearby tissues, but large enough that it doesn’t quickly filter out of the eye. Targeting VEGF 165 alone has proven to not be efficacious enough to reverse disease progression, companies now opt to block all VEGF-A isoforms, as some such as 121 and 189 have shown the capability to promote strong growth.
Wet AMD is classified into two main subtypes: Occult and Classic. Occult(Type 1) is when the CNV grows below the RPE and doesn’t break through. In occult wAMD the growth is contained under the RPE and progresses much slower. Historically patients with occult wAMD see less of an initial vision benefit after anti-VEGF treatment. Classic wAMD is the more aggressive and disruptive subtype. Classic CNV breaks through the RPE and causes much more structural damage. These patients see a much stronger visual rebound after initiating anti-VEGF treatment but are also worse off long term due to the structural disruption leading to photoreceptor death.
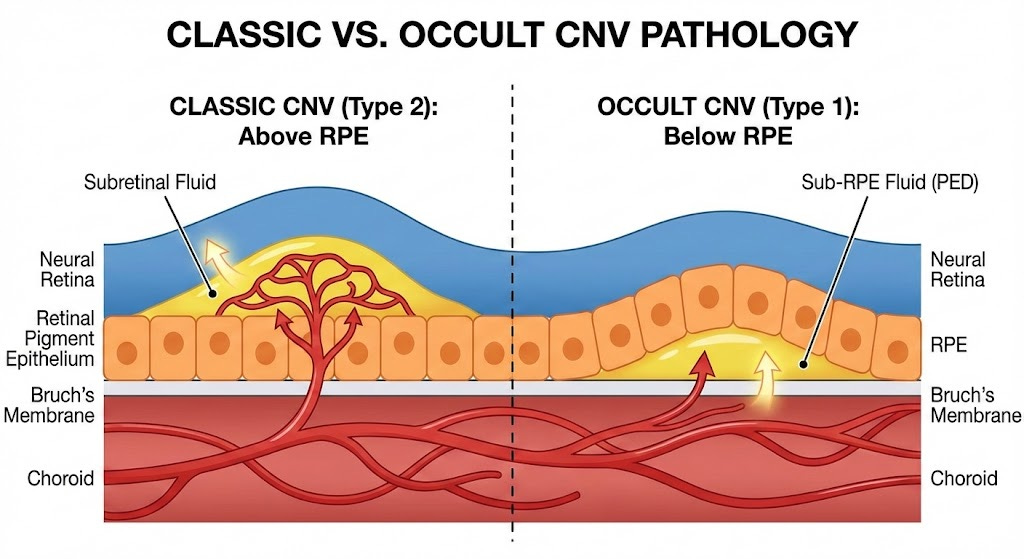
Source: Gemini
**Endpoints
The endpoints used in wet AMD to measure efficacy are primarily best corrected visual acuity (BVCA) using the Early Treatment Diabetic Retinopathy Study (EDTRS) chart and central subfield thickness (CST). BVCA is just a simple vision measurement with the standard 5 letters per line vision chart. You’re reading letters on the chart. CST is measured by the doc looking at your eye with an OCT (optical coherence tomography) and seeing the thickness of the eye at the macula.
**Market History
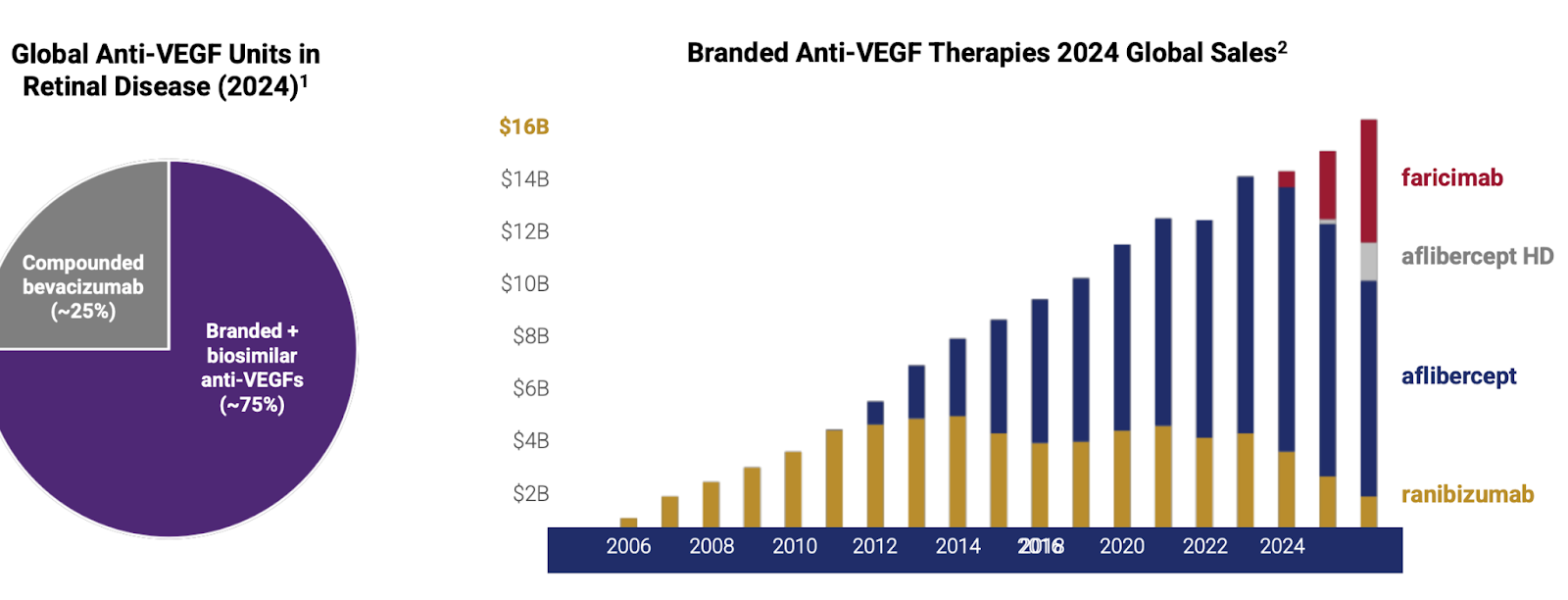
In 2004 the FDA approved Pfizer’s Macugen(pegaptanib), the first ever anti-VEGF for wet AMD. Macugen targeted the VEGF-165 isoform and showed a slowing of vision loss in patients of all subtypes of wAMD. Quickly after Macugen was approved, Genentech showed incredible data with their pan-VEGF-A fab, ranibizumab, demonstrating the ability to both shrink CNV’s, as well as meaningfully improve vision in patients with wAMD with Q4W dosing. Ranibizumab was approved in 2006 with the brand name Lucentis, and became a blockbuster in 2009 in the United States(Novartis held Ex US commercialization rights). Lucentis was able to achieve this market success despite competing with off-label Avastin, as quickly into the launch doctors realized they could substitute Lucentis with compounded Avastin at a much lower cost.
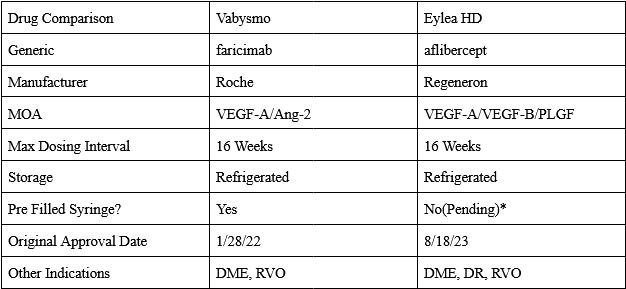
In 2011 Regeneron received approval of Eylea (aflibercept) 2mg for wAMD. Eylea is a VEGF-A and B ‘trap’ that acts as a decoy receptor. Eylea showed numerically equivalent efficacy to Lucentis with Q8W dosing, greatly reducing treatment burden. Immediately after launching, Eylea captured the majority of the wAMD market, reaching a peak sales of over $7 billion. They were so successful because of the decrease in treatment burden. Eye injections are uncomfortable and inconvenient, after seeing Eylea’s clinical data, there would be no reason for a doctor to use Lucentis. Eylea held the majority of the wAMD market share until the approval of Roche Genentech’s Vabysmo(faricimab) was approved in 2022. Vabysmo is a first in class bispecific VEGF-A/Ang2 antibody, and the first drug to be approved for up to Q12W(and eventually Q16W) dosing. Roche led an impressive launch for Vabysmo, facilitating the transfer of many from Lucentis and stealing market share from Eylea due to its superior product profile. The wAMD market is currently a duopoly, run by Vabysmo and EYLEA HD.
*Catalent lmao
The arc of wAMD bends towards DURABILITY
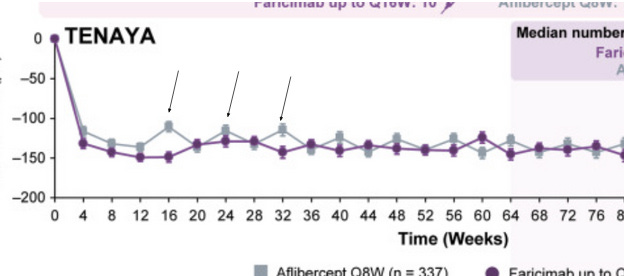
**TKI explanation
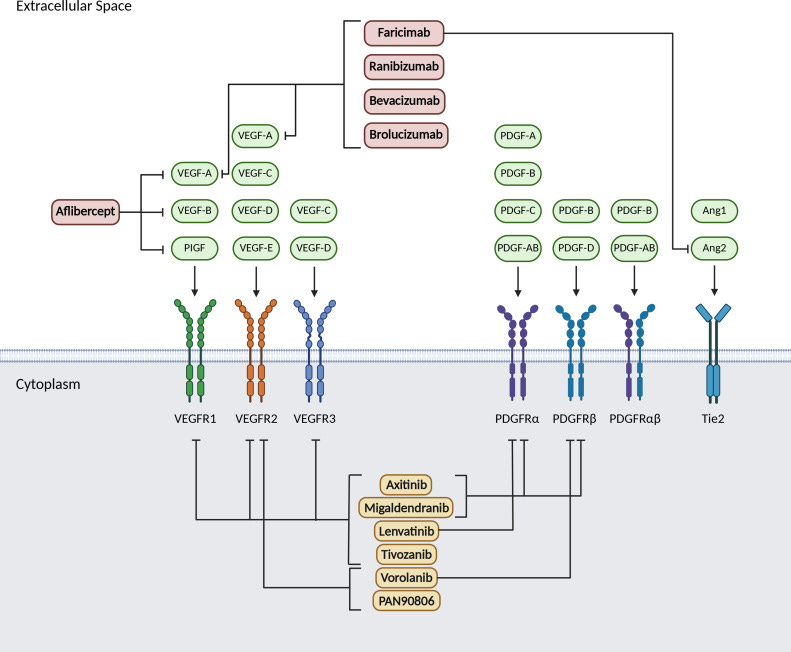
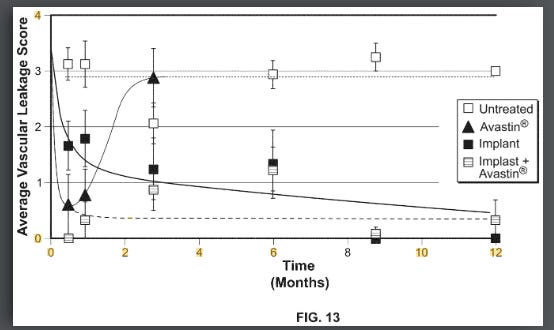
Ocular and Eyepoint are companies that are developing implants which secrete TKI’s in the eye with the goal of extending the wAMD dosing interval to 6+ months. These implants have the theoretical advantage over current antibodies by preventing the “seesaw” pattern of CNV growth in the eye by providing consistent strong VEGF inhibition. Well known drivers of worsening vision outcomes in wet AMD are fibrosis and macular atrophy. The constant healing of the retina and RPE after structural damage caused by bleeding from the CNV leads to a buildup of scar tissue and fibrosis. This scarring leads to permanent photoreceptor death, causing irreversible vision loss. Below is a chart of different dosing intervals which convey the “seesaw” pattern mentioned earlier via the central subfield thickness endpoint. Unlike the currently approved antibodies, extended release implants deliver strong consistent VEGF pathway inhibition until redosing, which should keep a patient’s CNV stable.
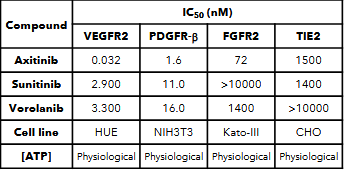
Ocular’s TKI implant (Axpaxli/Axitinib). Axitinib inhibits VEGF receptors at sub-nanomolar concentrations (in most assays), a chart of its selectivity is below. Axitinib inhibits VEGF receptors by competitively binding to the receptor’s intracellular ATP-binding site. There are three isoforms of VEGFR, and VEGFR2 (KDR) has the highest expression in the ocular endothelial cells and is the main disease driver in wAMD.
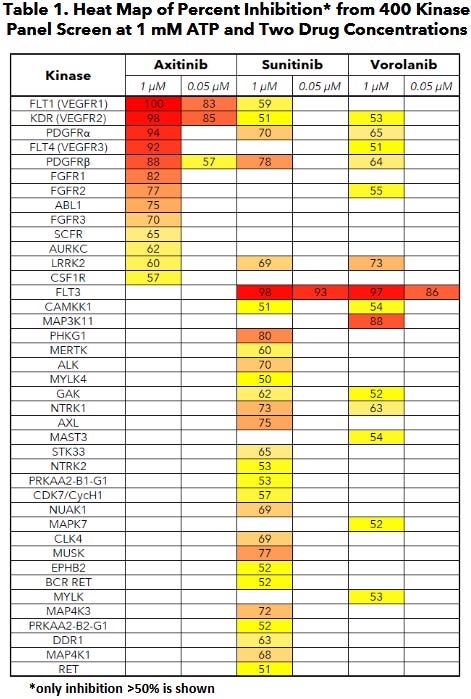
A few important things: this selectivity was determined by OCULAR (hence it’s biased) and assay dependent. But the potency & selectivity between isoforms between Ax and Vor are notable.
**History: GRAY, Graybug vision
We’ve tried this before with graybug vision and Sunitinib: GB-102 was a formulation of Sunitinib that failed on safety (drug delivery issue) and also showed Meh efficacy. Safety failed because the drug leaked into the anterior chamber, had a DLT in the high dose group plus many IOP incidents. But efficacy failed pretty badly: rescues at 4 months and loss of vision. It’s a knock on the TKI thesis.
https://pmc.ncbi.nlm.nih.gov/articles/PMC7000758/
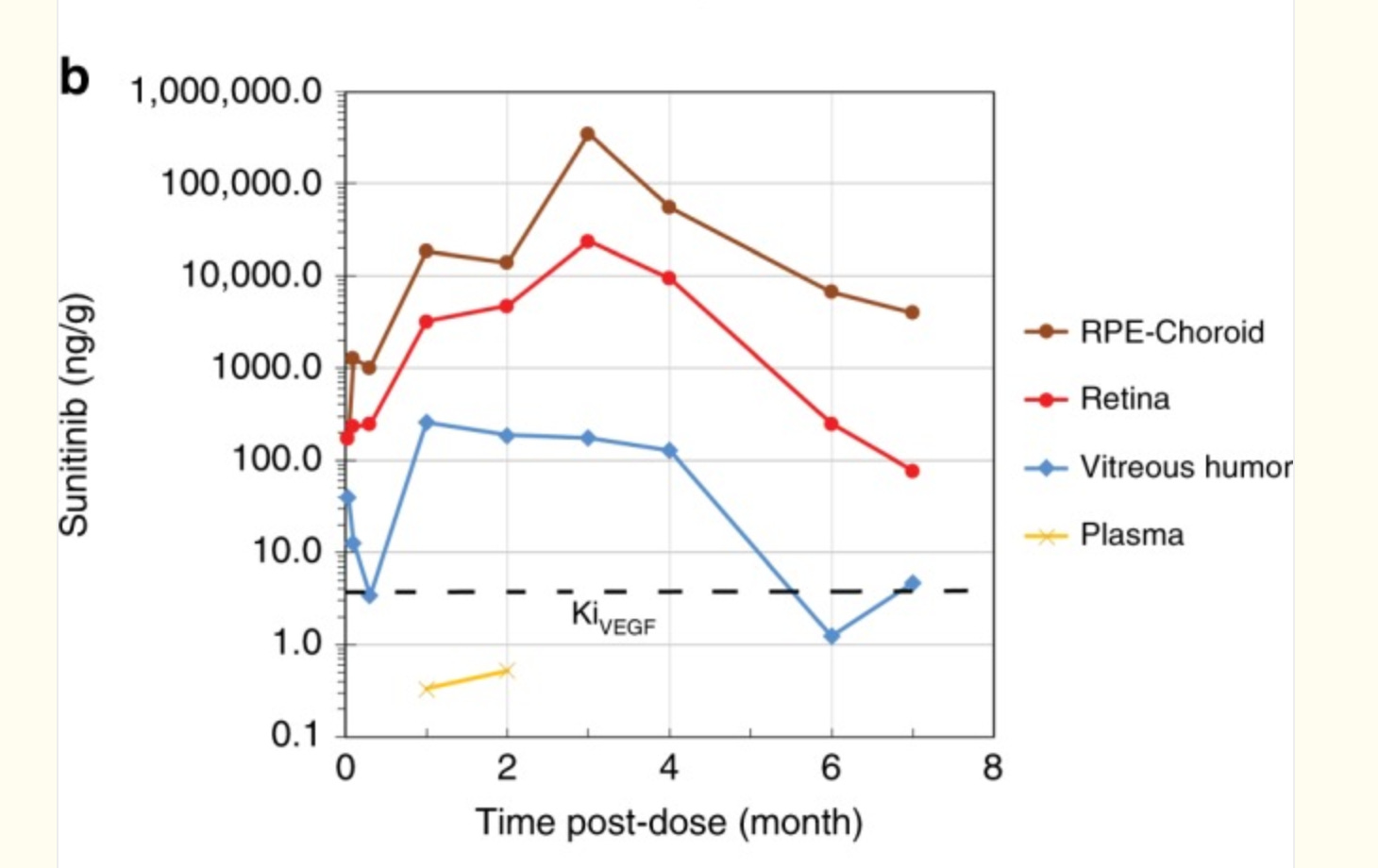
But whooooboy, there are some issues with this one. The release rate is Scurve, no 0 order so the concentration varies. In addition, Sunitinib is highly melanin-bound (80-99%)! The melanin-rich tissue such as choroid acts like a second depot, so it’s reasonable to infer that during the first few months of dosing, Sunitinib is just transferring from the original drug depot to the melanin depot with little free drug reaching the disease tissue. This corroborates with the continuous accumulation in retina and RPE/choroid in the rabbit PK study. The total concentration is so high, e.g., 500,000ng/g, that in the p2 study Graybug found a DLT in the 2mg group, raising safety concerns. Still, even with that, the rescues in phase 2a weren’t too bad. Notably better in those with less treatment burden up front.
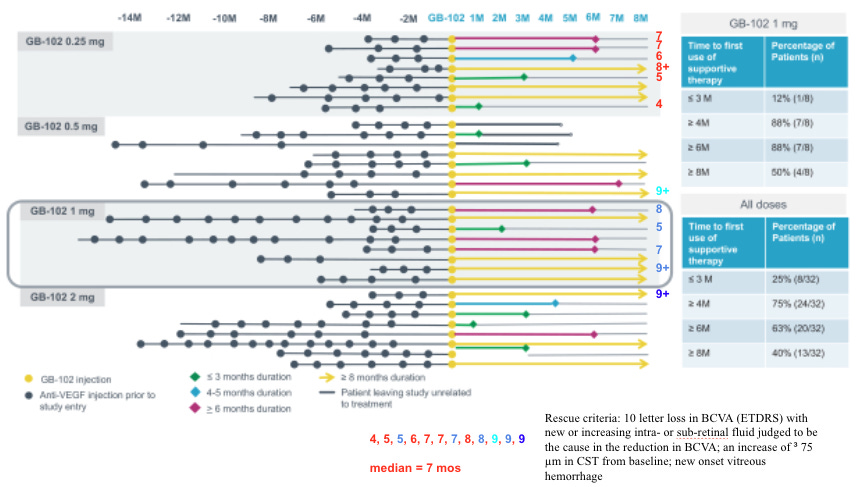
But the phase 2b was a fail: median time to rescue 4 months, worse on fluid. 7 letters worse.
Note: Axitinib and Vorolanib don’t bind melanin.
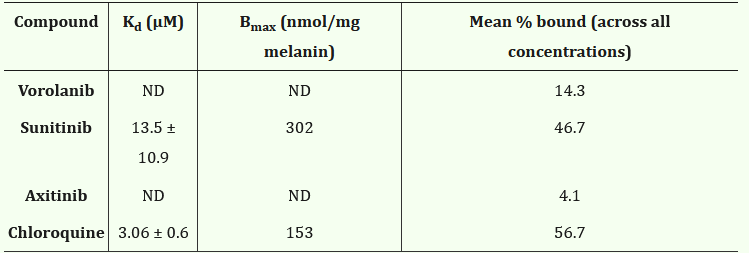
https://pmc.ncbi.nlm.nih.gov/articles/PMC11149885/
**why CLSD Axitinib “failed”
Before Ocular, Clearside biomedical tried Axitinib for wAMD. Clearside Biomedical developed a suprachoroidal space(SCS) depot of axitinib, but it didn’t match investor expectations. Clearside Biomedical’s clinical data wasn’t actually bad, but I think the complexity of this new SCS procedure made it a non-starter.
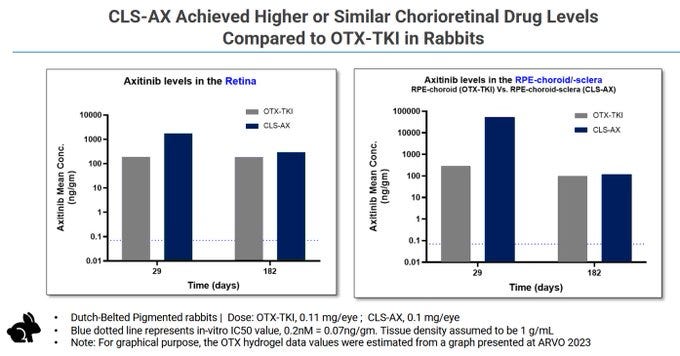
In theory, the concentrations are high out to 182 days.

But the other implants see deterioration at 90 days
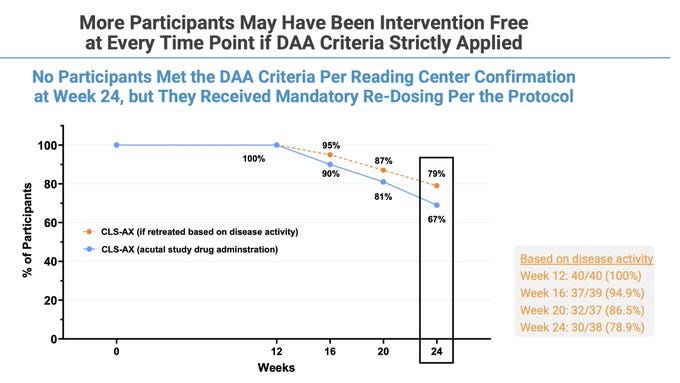
Rescue rates okay tbh even though “week 0” is before one more Eylea dose
The idea behind it is to inject a massive amount of axitinib crystal into the SCS, with the elevated pressure from the injection helping to push some of the crystal into surrounding tissues. The crystal dissolves over time allowing for free drug to act on VEGF receptors. This method seems to lead to quite a lot of randomness. In wAMD there is a degree of difference between patients when it comes to RPE structure and permeability so the diffusion of drug can impact local concentrations. I have a hunch (and no I’m not opening consol to model it) that the local concentrations vary even if total concentrations are high. Since SCS injections are much less common than IVT injections, any imperfections aren’t tolerated.
Importantly, only “treatment experienced participants with reading center confirmation of persistent active disease” were enrolled into the trial. The patient population was experienced.
The CST and BVCA curves show the same trend as other TKIs, numerically slightly worse efficacy than aflibercept, but within error. So I have a theory: they need $ to fund a ph3 as an equal/slightly worse competitor with a weird dosing method. Clinical data is OKAY but not great, and SCS procedure has room to improve. But it ends up a 0.
**Back to OCUL: PK and PD
Historical literature on axitinib has indicated that its IC50 for VEGFR2 (KDR) is 0.2nM, although Ocular has shown in an in-house study that at physiological levels of ATP axitinib’s IC50 is 0.032 nM (below, I don’t have good intuition where they derived this IC50). Based on the bulk of independent studies, it’s reasonable to believe the IC50 against KDR is in the 0.2-1nM range. Very potent per se.
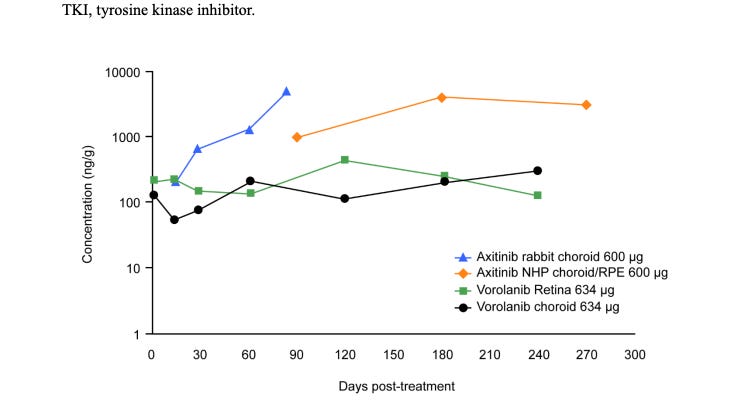
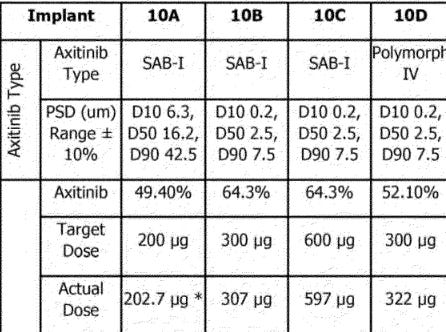
Initial PK results. These results are NOT the same as the one used in the final Ph3. Ocular developed a NEW implant (Form IV in their patents) with a different, more consistent release profile. A few notes before the patent data.
Rabbit Vitreous fluid is 3X smaller than humans (1.5 vs 4.5 ml)
Zero Order kinetics = release is roughly a function of the number of implants but not dependent on drug remaining (before the matrix breaks)
Rabbit drugs clear drugs faster, requiring ~50% more drug to be equivalent.
IC50 = .03nM. 77 ng/mL = IC90, hill coefficient 1 (assuming 1% free drug)
The phase 1 trials used the older SAB-1 Formulation. The SAB-1 design released less drug shortly after being dosed and had a large bolus of drug release at the end of the implants life. The new Form IV formulation fixes these problems. It releases drug quicker than SAB-1, and shows more consistent tissue concentrations over time. The SAB-1 data displays massive variance, it seems that some of the implants secreted almost zero drug at the first timepoint. The goal for the FORM IV is to be BETTER
“Well, we would expect that number to be the same or higher. I mean this new formulation will deliver just like the older formulation or the pre-optimized formulation. It will deliver drug continuously for 9 to 12 months. And as we mentioned for the first 9 months, it will deliver drug at a slightly faster rate than the 600 microgram dose. So up until 9 months, you’re actually going to get more drug to the target tissues than you would have with the previous 600-microgram formulation. So up through that 9-month time frame, we’re very confident that we’ll do at least or better than that.” 11/2023 conference.
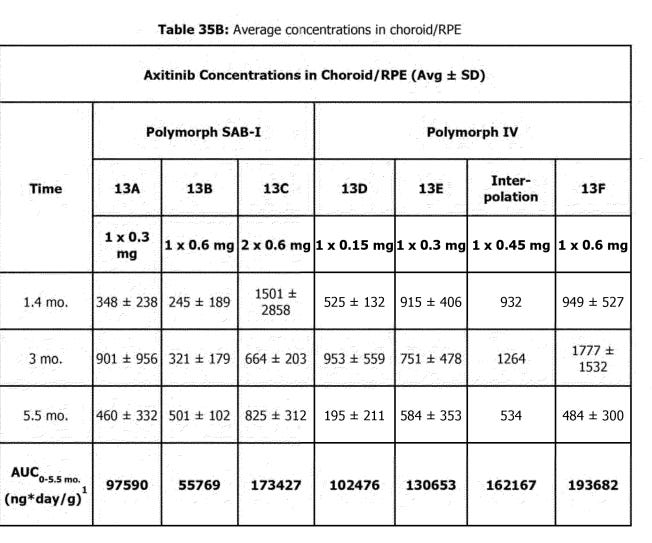
SEE: SAB- 1 SD = 2X mean at 1.4 mo.
The theoretical benefit of the higher AUC and more consistent dosing was shown clearly in a rabbit VEGF challenge model. This model works by injecting a highly supraphysiological dose (1ug per eye, literally cancer causing dose) of VEGF into rabbit eyes and measuring vascular leakage. Form IV beat SAB-1 AND showed persistent control.
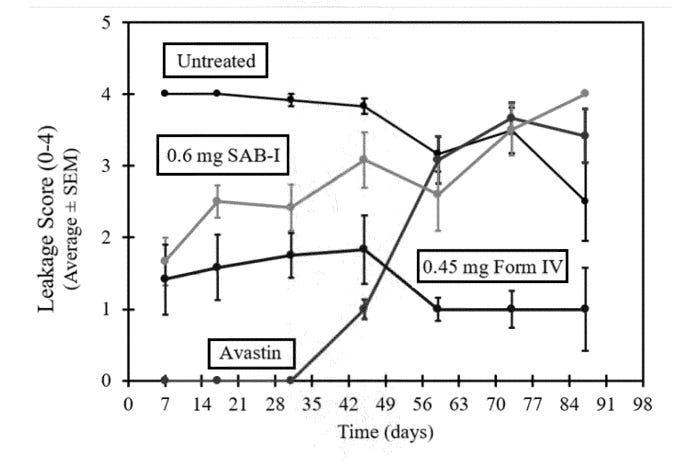
Form IV patent
There has been some discussion about how the axitinib based drugs did not match the efficacy of bevacizumab in this test, it is important to put into perspective that:
There were no anti-VEGF loading doses in this test for the axitinib arms like there are in their RCT
The dose of VEGF injected into the rabbit eye was 1 ug (1,000,000 pg), the standard concentration of VEGF in human eyes is in the X00 pg/mL range VEGF in wAMD . We’re injecting a cancer-causing dose of VEGF into these rabbits. I think it’s because 1000X the VEGF in human eyes has off target receptor binding to NRP1.
Many of these graphs shown have been from patents filed by Ocular. Seen below is retinal concentration data from 3, 6, and 9 months in NHPs:
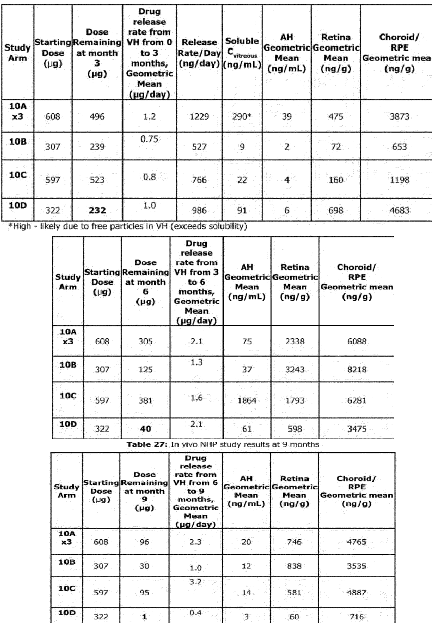
The NHP study continues to show that SAB-1 does not release enough drug initially after dosing and that the Form IV design allows for relatively stable concentrations of axitinib in retinal tissues. One crucial detail is that this study used a 300ug implant for the Form IV arm, and the current in human phase 3 studies use 450ug. Due to the design of the Form IV implants aiming for near constant rate release, this data indicates that the 450ug dose should last >7 months in the eye, and up to even 9 months.
Due to the lack of initial drug release in the SAB-1 arms and the large variance in all SAB-1 concentration data, it appears that many of the underwhelming responses in the in human data could be attributed to a lack of suppression soon after dosing, allowing for disease to progress and become harder to control.
Note: in Fig here, injecting 1ug VEGF IV and even SAB-1 shows durability out to a year.
Axpaxli Clinical Data
Ocular has run two prior phase 1 trials, each with an n~20. Management has changed the formulation during development, this trial utilized the obsolete SAB-1 design. Data looks alright.
The Australian phase 1 was the first in human study of Axpaxli and displayed clinical activity at all dose levels. There were no SAEs attributed to Axpaxli and the 6 month rescue free rate was 60%. It is notable that 1) some of the cohorts didn’t receive any aflibercept loading doses; 2) the population is a mixture of treatment-experienced and -naive; and 3) that many of the supplemental aflibercept doses did not meet the prespecified rescue criteria.
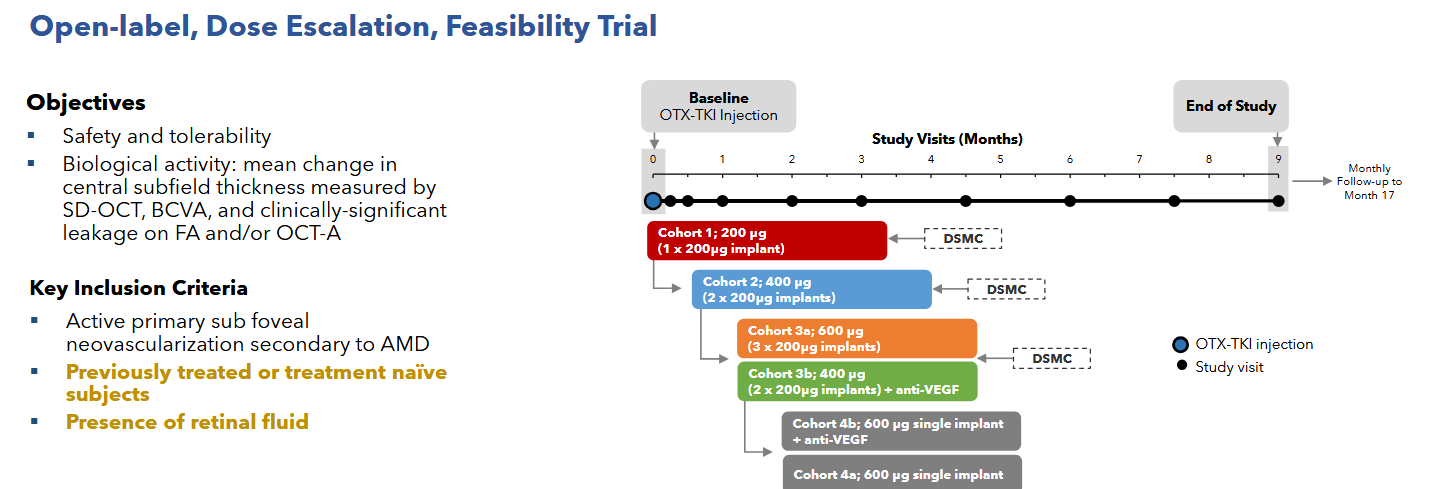
Notably, part of the inclusion criteria for this study was that a patient must have retinal fluid presence, implying that the patients enrolled in this study had active disease.
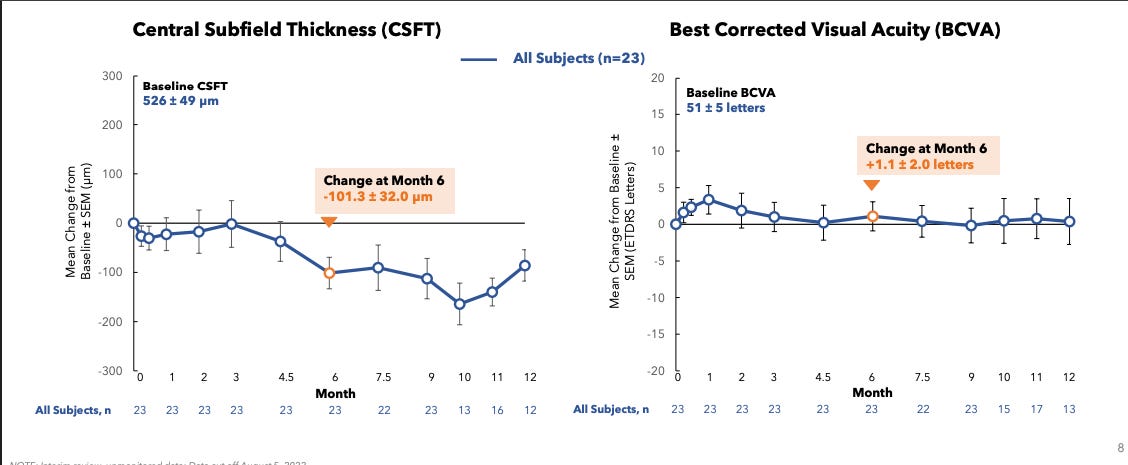
Patients also were, as the kids called it, cooked. Baseline fluid was 526 and BCVA 51.
There was one case of intraocular inflammation in cohort 3b, it is unclear whether that was due to the Axpaxli or aflibercept injection. The company never released data from the fourth cohort in this trial, presumably the 1 insert of 600ug with the old formulation failed to control disease in these more aggressive disease state patients. It’s worth noting that even if the patients were much worse off and had active disease and no loading doses, we saw good signs of efficacy especially in the treatment naive population. In Cohort 3a 4 out of 5 treatment naive patients with active disease maintained a good status for >=6 months without rescue & without any loading anti-VEGF injections.
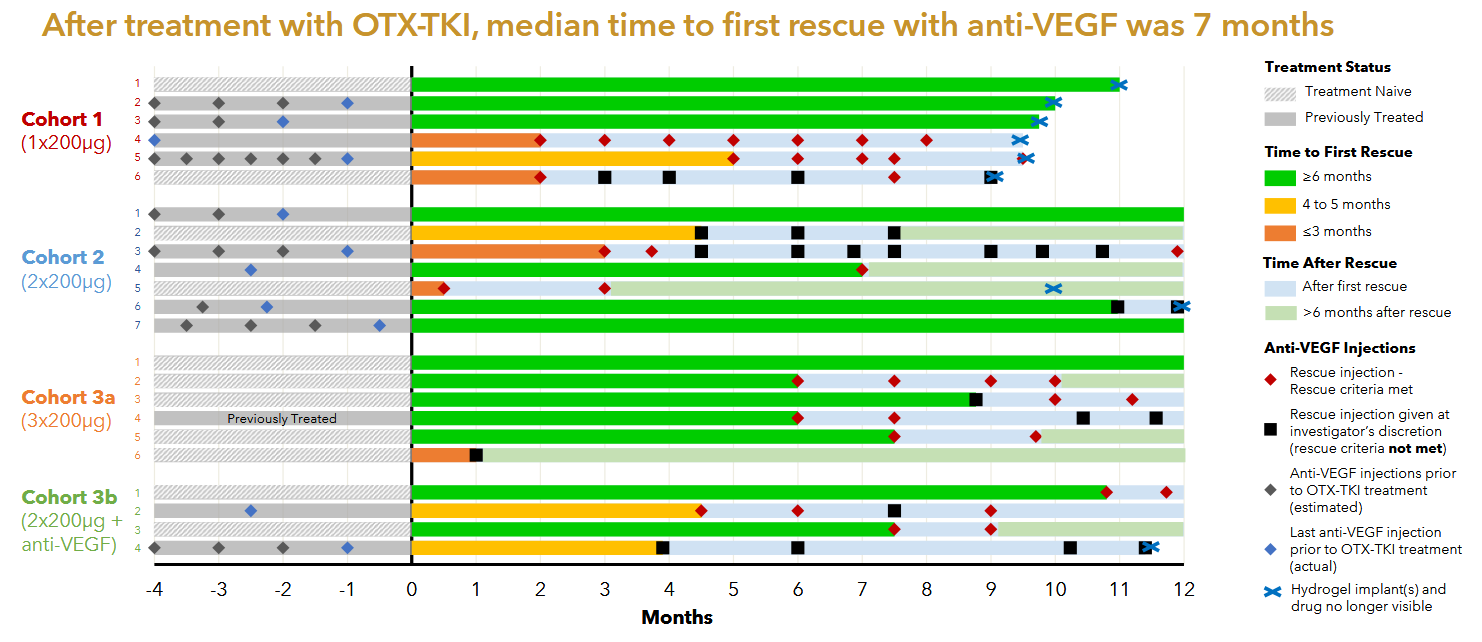
US Phase 1
The American phase 1 study is more rigorously designed, as it included a control aflibercept arm and had an inclusion criteria much closer aligned with SOL-1. The American phase 1 was randomized 3:1 to either a 600ug Axpaxli injection or Q8W aflibercept. This study also used the original SAB-1 formulation. Interestingly, the loading aflibercept injection in the Axpaxli arm occurred after the Axpaxli injection. This is suboptimal, it makes much more sense to rapidly dry the eye with aflibercept, then dose a TKI after to block ATP on the receptor for preservation of anatomical response. This lesson was learned for their phase 3 trials.
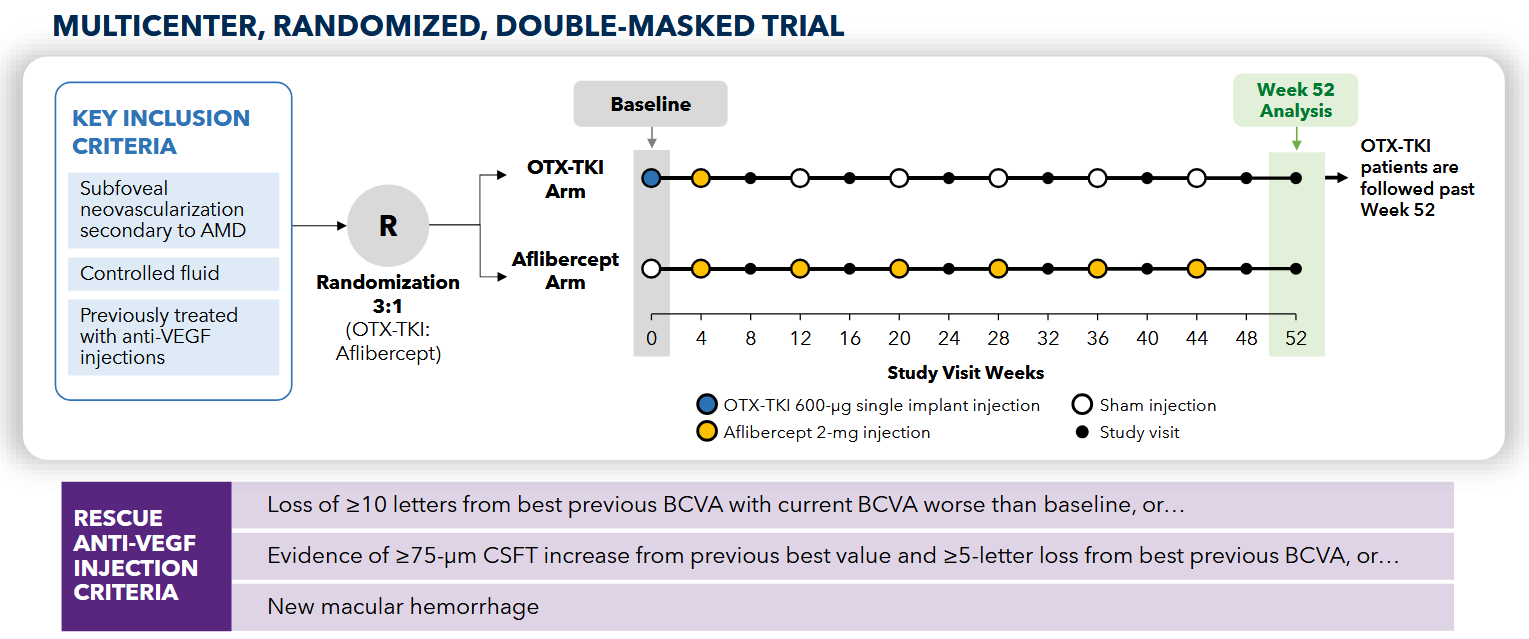
The trial uses a little different design because lower baseline and get one dose eylea right after implant. Here, I’d estimate ~3 patients required per protocol rescue at 40 weeks. Two patients get non-protocol injections and don’t need anything else for a whole year (10,13). I suspect this is an artifact from the old SAB-I formulation, as it releases the drug slowly at first, then spikes upon bioresorption ~6-9 months. The discontinuation of rescue coincides with this release kinetics. Remember, this is the old formulation. The new formulation is designed to be more zero order which should mitigate the low exposure issue in the first few months.
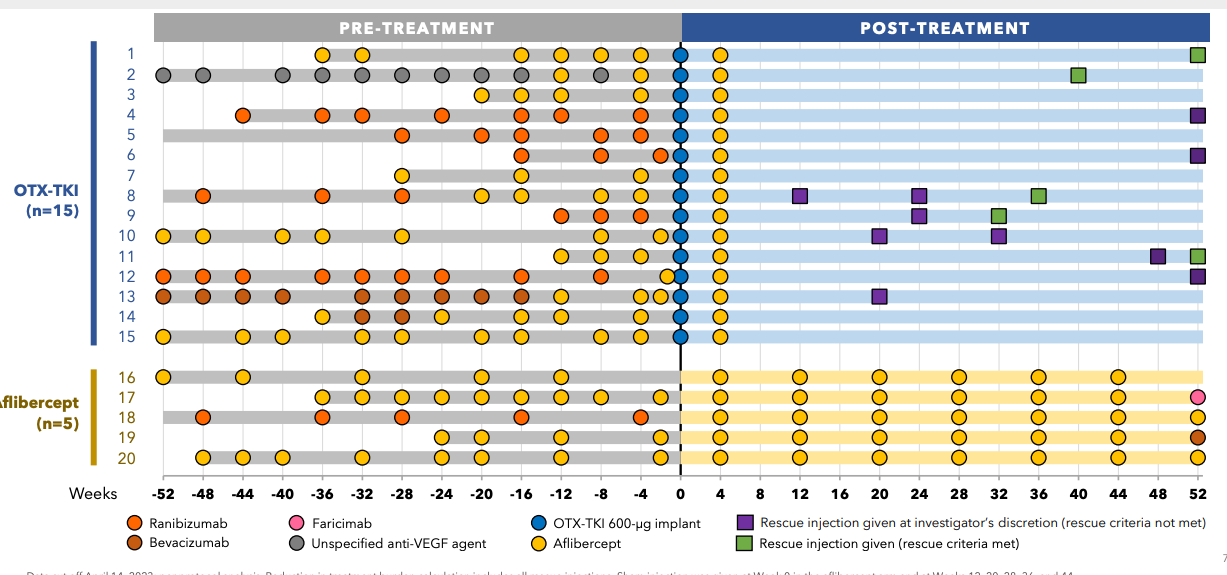
**SOL-1 - rigged game
SOL-1 is the main catalyst. And they’ve clearly rigged it for success.
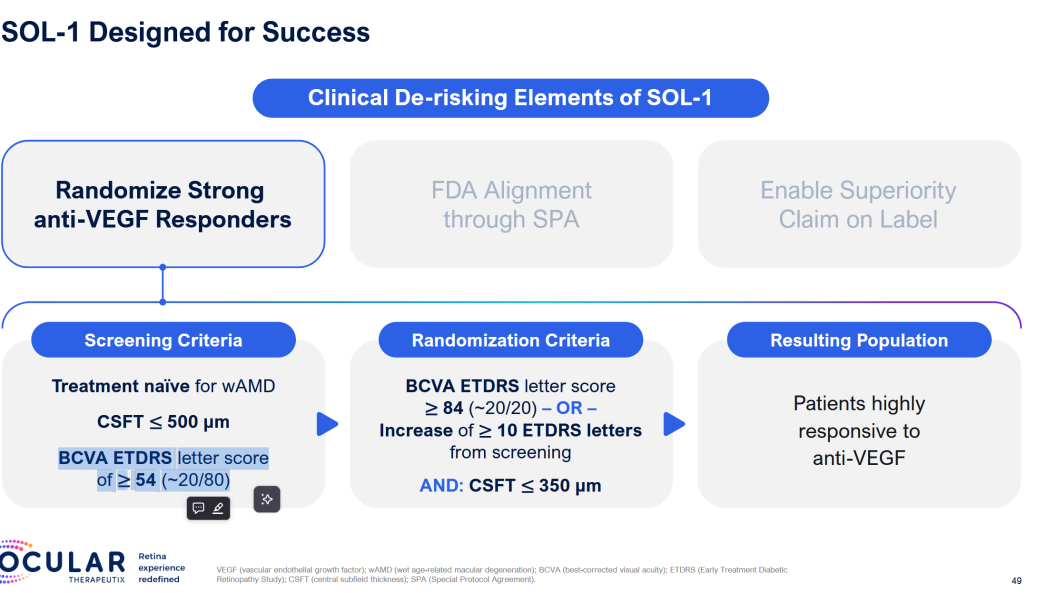
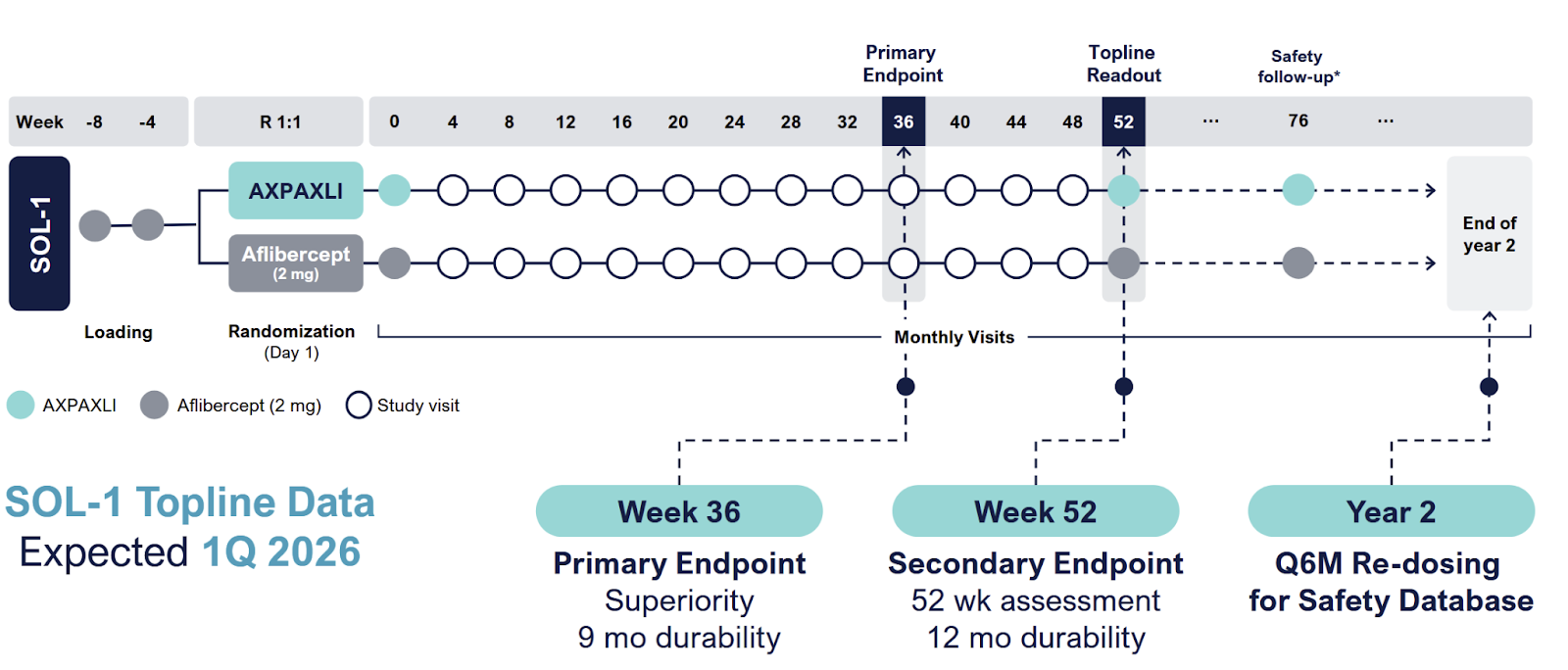
Patients get two loading doses, they HAVE to improve, then they get Eylea or Axpaxli, and they check for superiority at 9 and 12 months. They are selecting for patients who A) respond and have B) mild disease. We want to see if 1) these patients require less than expected VEGF (as TKI < VEGF) and 2) still require adequate rescues at 9 months based on vision loss. The primary endpoint is loss of 15 letters from the randomization baseline (note: it is the “peak” baseline after the patients respond to the two loading doses; not from the screening baseline; so essentially the primary endpoint is the gross loss of ~5 letters from the screening baseline).
The trial is initiated under an SPA to prove superiority at 9 months. Total 344 patients,
Why the 15 letter loss?
Straight from FDA guidance:
The sponsor should enroll patients
Who are expected to decline or have lost vision
Primary endpoint is more patients who gain 15 letters, less who lose 15 letters, or a delta of 15 letters.
Same treatment regimen between drugs. (the Vabysmo review calls this out)
Ocular enrolled 344 patients, and in March 2025, extended the blinded period out to 1 year to enable redosing and shrink the other pivotal SOL-R study. More patients, more safety data. Data for SOL-1 is expected in February
With this trial: they’ve done a few things
Enroll patients who presumably require lower anti-VEGF injection frequency
Still will likely fail <9 months
Choose an endpoint the FDA recognizes as ‘approvable’
Choosing patients who respond
Vabysmo Phase 3: trial
As more durable anti-VEGF antibodies have joined the market, doctors and investigators have discovered an interesting phenomenon: Those who have the best vision gain from initial VEGF therapies benefit the best from extended dosing antibodies. Faricimab and Tarcocimab both demonstrated this in their prior phase 3 PRN trials.
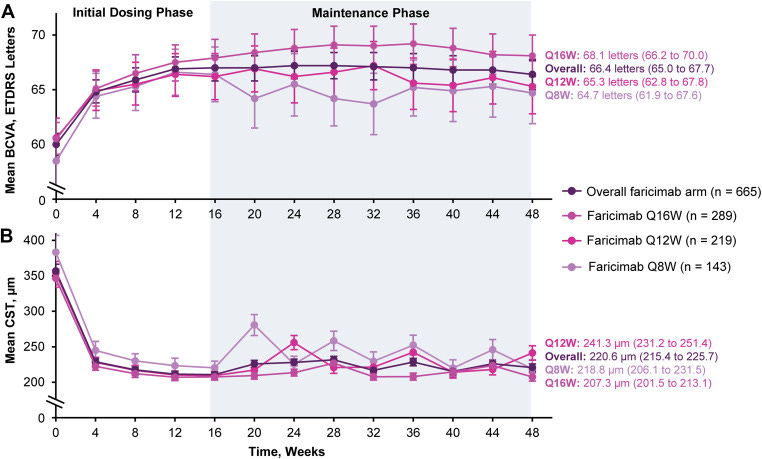
Those who needed more frequent dosing had a lower visual response to the initial doses to anti-VEGF. The PK of faricimab extends out to~12 weeks so the Q16 dosing must mean these patients don’t require active VEGF suppression!
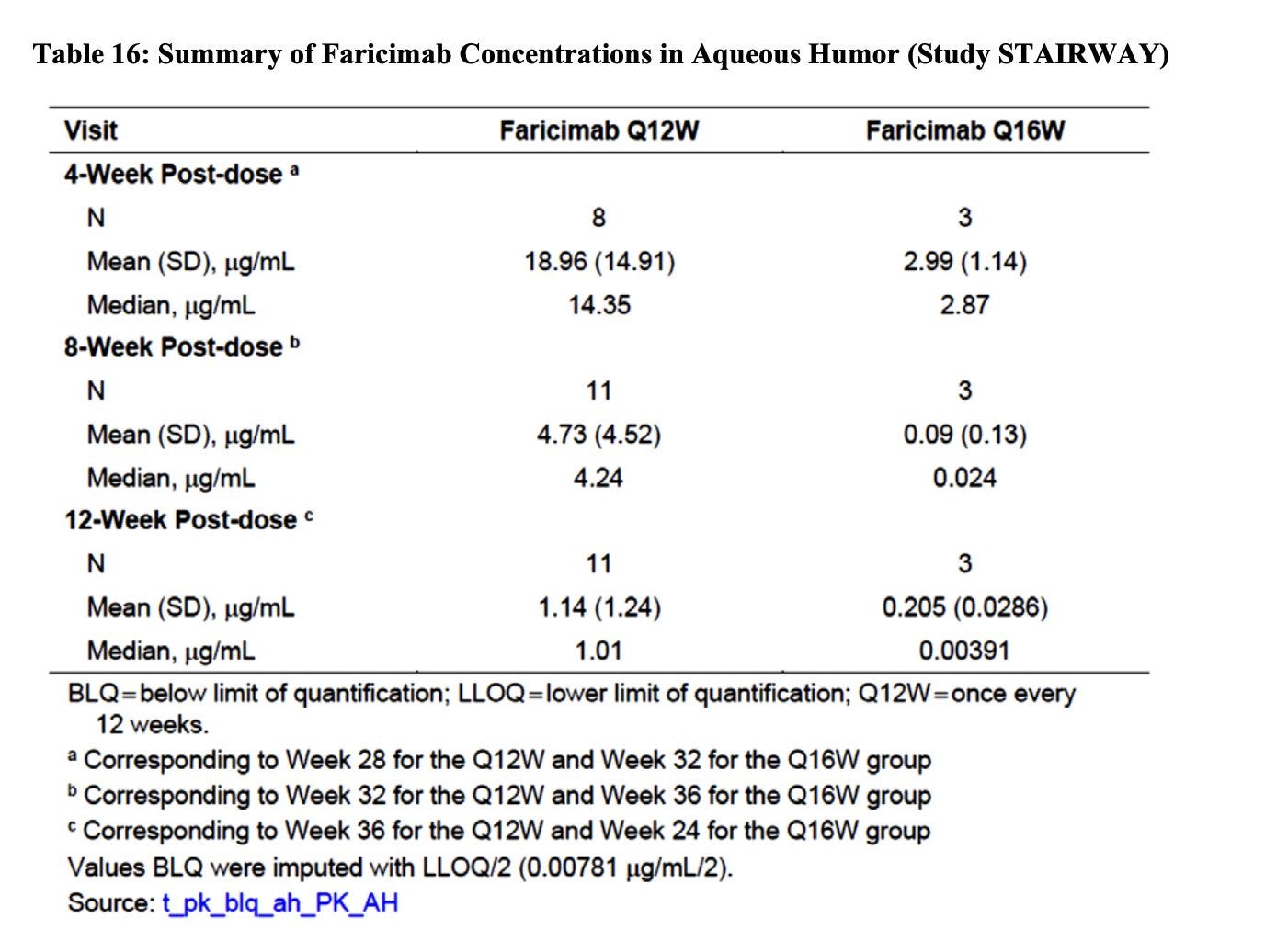
Kodiak Patients: their ph3 trial failed but provides a view into the patient subset that responds.
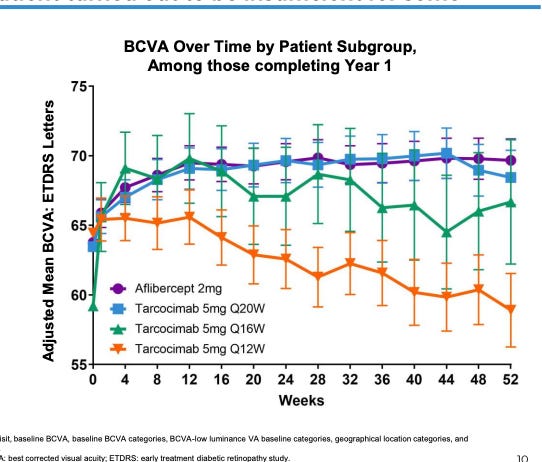
Patients who needed > Q12W dosing had barely any initial response.
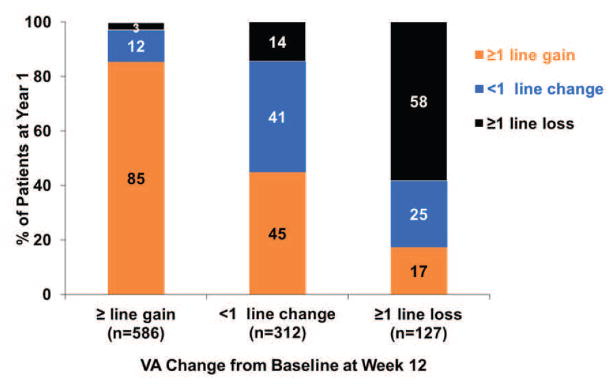
CATT trial: A treat and extend type model.
Patients in this trial were allowed to switch to PRN drug if disease activity was sufficiently suppressed. Here, consistently, patients with initial visual gains were able to extend the trial. Obviously confounded by how initial gains allow patients to maintain the gains, but 80% of patients with initial 3 letters were able to maintain it with VEGF treatment. If you can suppress these patients and they gain letters,they’re likely to maintain that gain. And this is maintaining 15 letters of vision, not prevention of loss. +1 for easier to treat population
Obv confounded because it’s real world data
One last one for the road: A metaphor for all of these.
But what about the rescue rate? What if that outperforms too?
The flipside of treatment arm OUTPERFORMANCE is control arm outperformance: what is the expected rescue rate in the control arm?
Zoom out for one second: if I gave you one dose of Eylea, what is the chance you won’t need another dose for 9 months? The answer is 0. We’ve never tried it before so we don’t know with certainty, but in the real world, patients very rarely get 1 dose per year. However, it’s not the 1 dose criteria but rather losing 15 letters in 9 months which matters.
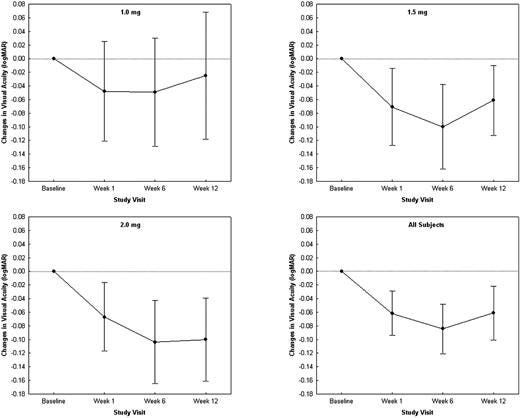
Phase 1 Dose escalation trial: Baseline 20/320 vision.
Here, the patients get one dose of Eylea, gain about .1logMAR (5 letters) and maintain out to week 12. In the 2mg group, we don’t see a return to baseline but we do see in the 1mg group return to baseline likely around week 20? Error bars too large to interpret and baseline is awful vision…..
\
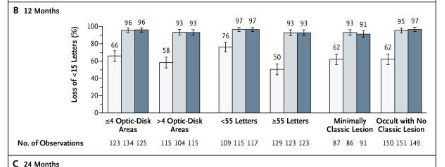
Ranibizumab vs Sham: baseline 53 letters, predominantly occult lesions. 63% of SHAM injection didn’t lose 15 letters at baseline
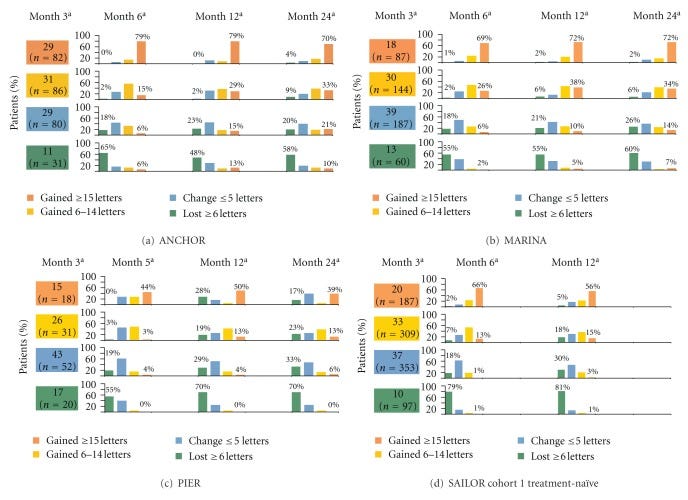
Patients with treat and extend therapy are monitored for a few months after stopping therapy. Patients with AMD were treated with 3 monthly injections and then followed in a TEregiment with extensions by 2 weeks. Patients reaching 12 weeks WITHOUT any disease activity on 3 consecutive visits were eligible. Then we track for recurrence.
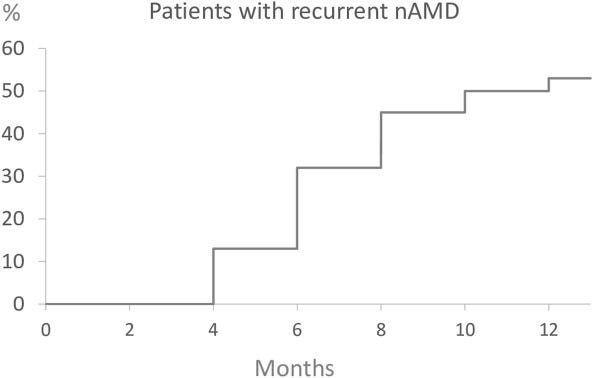
Hmmmm, Only about 50% have a recurrence at 9 months?
I don’t think this trial represents the patient population and represents the best case scenario. Only 40%of patients actually reach these criteria for extension to 12 weeks. These patients were slowly extended over time
Verteporforin vs placebo 1: mean 65 letters in study (also interesting 20 letters more than the other eye….)

% who lose 15 letters. At month 24, in the placebo arm, oddly it’s lower in the better vision patients (still 63%+).
Here we see this: patients with better vision have ~34% without loss of 15 letters. Patients with baseline vision < 54 letters have a 40% chance to survive the 15 letter loss in a year. The delta is more pronounced in treated patients. Note this is at month 24.
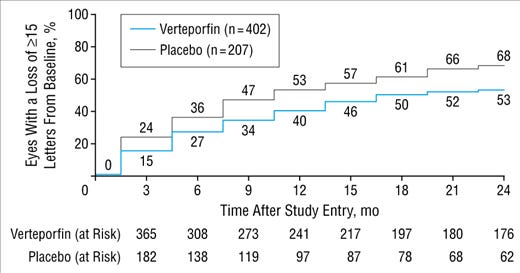
If you look at the actual curve, ~50% and 34% of placebo and verteporfin lose 15 letters.
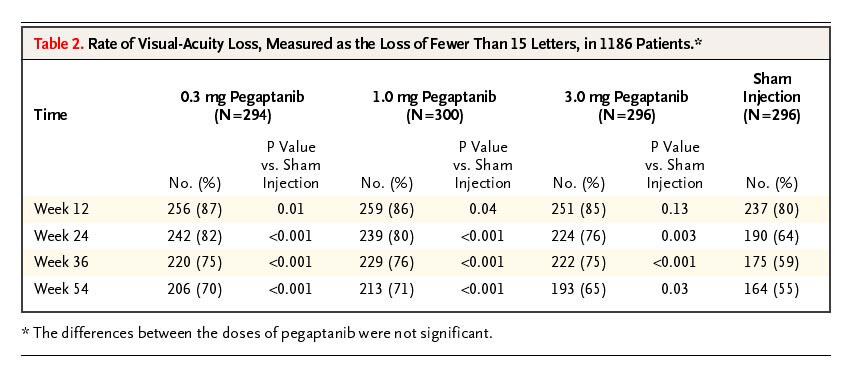
Approximately 40% of placebo don’t lose15 letters at 9 months.
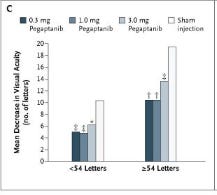
Again, at one year, about 15 letters lost in the sham arm and it’s ~20 in the One with baseline > 54 letters (OCUL trial).
So we have 4 data sets with placebo or no control treatment data. Across most cohorts (save for one verte group), patients with better vision lose more vision.
Ranibizumab - 50% >55 letters lose 15 letters at 12 months
The treat and extend data - of the 40% that reach 12 weeks intervals, ~50% recur at 9 months. On average they’ve lost < 10 letters at recurrence.
Verteporforin - 47, 50% lose 15 letters at 9 months, 36% at 6 months.
Pegaptanib - Overall 40% lose 15 letters in the sham at 9 months, 35% at 6 months. Likely higher in patients with > 55 letters at baseline.
Further, the PK of eylea has an ~11 day half life in the eye. To be on the safe side, let’s assume it goes to 0 in 3 months. Now: assuming the drug is active for 3 months, and inactive for the next 6, using the sham controlled data, across multiple cohorts, only 35% would lose 15 letters! That’s an issue because 1) the stat sig is called into question (OCUL guided to 70% rescue rate in the Tx arm, 20% in the control arm) 2) what if we used HD eylea instead? But even in that case, the drug can still potentially be worth 1B-2B (i.e. flat stock price) as a foundational treatment with a longer dosing interval on the label.
But that is the bear case scenario using assumptions which are not correct:
Patients with better vision lose more vision. The 35% placebo rescue rate at 6 months is in all cohorts, Patients with > 55 letters at baseline tend to lose vision at ~1.5-2x the speed. This is likely close to 50% in that subgroup
We are selecting for patients who initially improved in vision. The true baseline for these patients is not at month 1 post 2 injection Eylea. It’s at month -2, before the eylea injections.
The PK profiles out to 3 months is generous. The half life is 11 days and modeling shows suppression out to 71 days
The most important: real world data.
I think this real world data provides us information on the efficacy of eylea. First off, patients with better vision lose more vision, consistent with the prior data. The <7 anti-VEGF group is better suited for comparison to the SOL-1 population as they presumably require less VEGF suppression. (interesting there is minimal difference in baseline VA and requirement for injections.
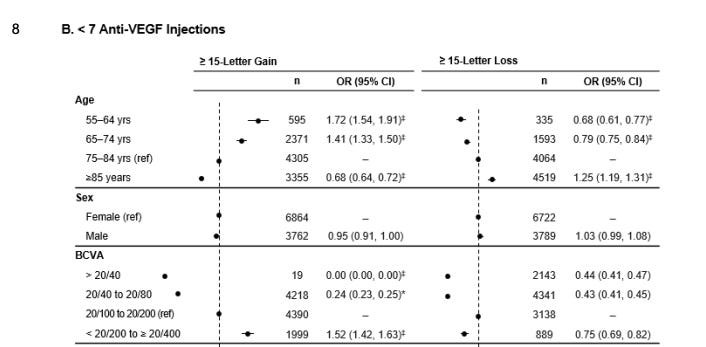
Same here, the baseline rates of loss of 15 letters
But here’s the interesting part: we can bracket a floor and a ceiling from this data from the subgroups who require less than 7 injections.
What % of patients are receiving < 3 injections in a year? This is the ~ group for patients who wouldn’t need a rescue.
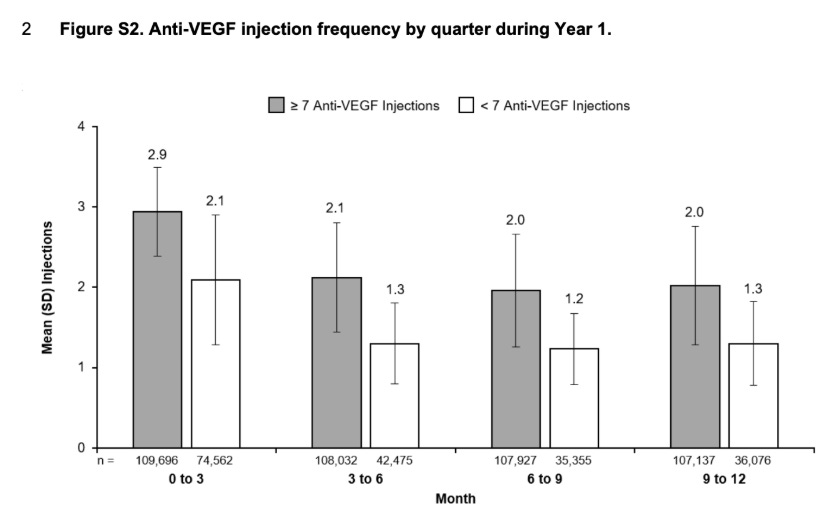
Plug in Mean, SD of injection frequency into chat GPT and ask for a distribution: approximately 40% of eyes with < 7 injections required < 4 injections in the year. This is a conservative estimate still. Because the distribution may vary. It’s not the Percent

Furthermore, What % of the individuals receive injections only in Q1,Q2, none in Q3, Q4 (estimating those without a rescue later). This analysis does not reflect real world practice, It’s kinda nonsense. But I think it is still useful because it places a floor on the control arm. ~30% of patients don’t need a Q3/Q4 injection

What % of GOOD vision patients still lose 15 letters even though they are presumably suppressed?
About 12% of patients lose vision even with adequate suppression (in theory). This is the peak durability.
Estimating the Control Arm, Treatment arm:
Putting all of this together I estimate about 90% vs 50% rescue rates at 9 months.
Across trials - about 30-40% loss vision at 15 letters at 6 months in the sham arms. The HR is ~.5 for patients with vision > 55 Letters. Here, we are selecting for patients who lose vision slower, but gain vision when on drug.
Assuming 6 months drug free, equal balance patients who respond, aptients with better vision, I get to 40-50%. I add on a few for non protocol rescues.
10-20% of patients just lose vision thus Axpaxli won’t be able to prevent rescue in those patients. If the pharmacokinetics hold, 0% of the patients should require a rescue. A small discount for the expected rescues in refractory disease.
None of the patients in the phase 1 trial lost 15 letters.
The Prior formulation is worse than the current formulation.
**SOL-1 Bar/readthroughs
Bear case - Fail - 10%
Base case - stat sig - 70 vs 30% delta, up 30-40%.
Bull case - 80%+ rescue free rates at 9 months. -> should be up 100%
Expectations set at 70% vs 20%.
Can you extrapolate this data to their non-inferiority trial and confidently assume success?
The NI study redoses at 6 months (SOL-R, compared to Eylea 2mg Q8 and Eylea 8mg Q6, interestingly EYPT doesn’t have a Q6 arm even though guidance asks for it….). The most important part of this trial is not the BCVA. It’s the CST. A dry eye is a good eye. If the CST is rising, there is excess fluid in the eye and the disease is progressing. As long as the CST curve is stable we know the drug is working and the disease is not progressing. The trial is a success as long as the CST doesn’t begin to show an aggressive rise before the 6 month mark.
Furthermore, SOL-R is enrolling patients without persistent fluid after Eylea. Those patients will screen fail. Consistently across trials, patients with no IRF do better.
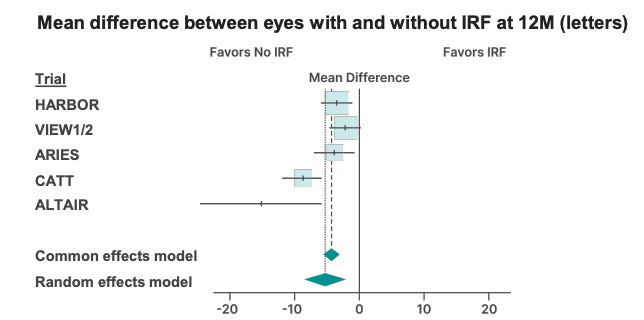
Is the delta on the primary endpoint sufficient to convince the FDA to extend dosing past 6 months on the label and/or file on 1 trial?
Opthals ignore labels, but insurance companies do not. Plenty of prior issues when eylea came out without Q4Wdosing so it couldn’t get reimbursed. Here, I don’t know how they’d approach a “superiority label” but we won’t know until the data comes out. The bar you’ll probably hear about the trial is 60-70% rescue free at month 9 and reaching statistical superiority compared to the control arm. This is quite a good idea for the bar, if docs can hear that they can put the majority of their new patients who respond well to the first VEGF treatments on an implant that gives the doc ample time between visits, the drug will get good uptake.
Failure? Stat failure is at <15% delta. I view it as unlikely.
**Safety
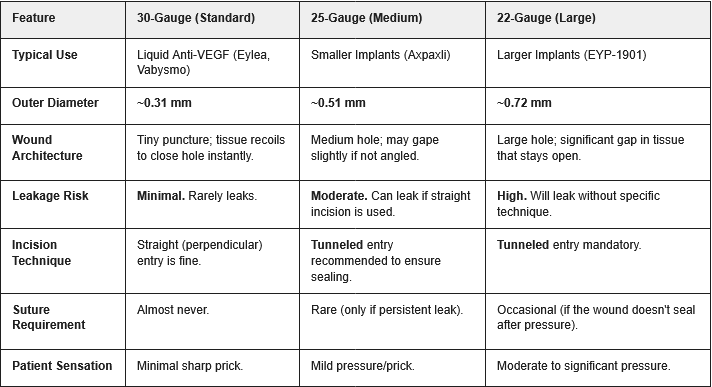
We saw one case of inflammation so far with an aflibercept injection in the phase 1. I have no way to handicap the safety for the new formulation. One thing I’ll note is that EYPT is using a 22 gauge needle, Ocular 25 gauge needle and Eylea a 30 gauge needle. EYPT’s needle size is larger than allows the eye to self heal. Keep an eye on any IOP elevations in that trial with chronic injections.
Comp to EYPT:
All these words just say “buy the TKI class”. But it’s not that simple. EYPT’s drug is not as good. They’re saying it inhibits IL-6 (which does work), but that ain’t it dawg. They do not hit IL-6 imo.

All in good fun, they may still do well.
Furthermore, they’ve shown some preliminary data that dosing >>>>IC50 doesn’t matter (because they do not dose >>>> the IC50), but that paper has a host of issues: the CAM sponge assay doesn’t represent a real VEGF in the eye model and they’ve given 200x the amount of vorolanib compared to axitinib (because the IC50 is 200x less!). In the model, VEGF performs worse which we know is likely not true.
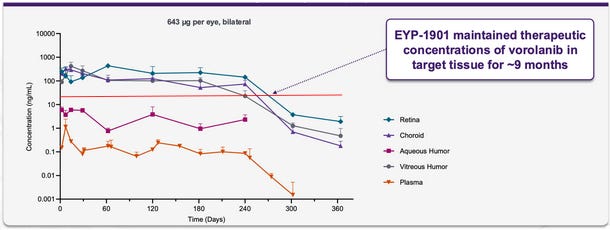
Red line is IC50 at 23 ng/mL
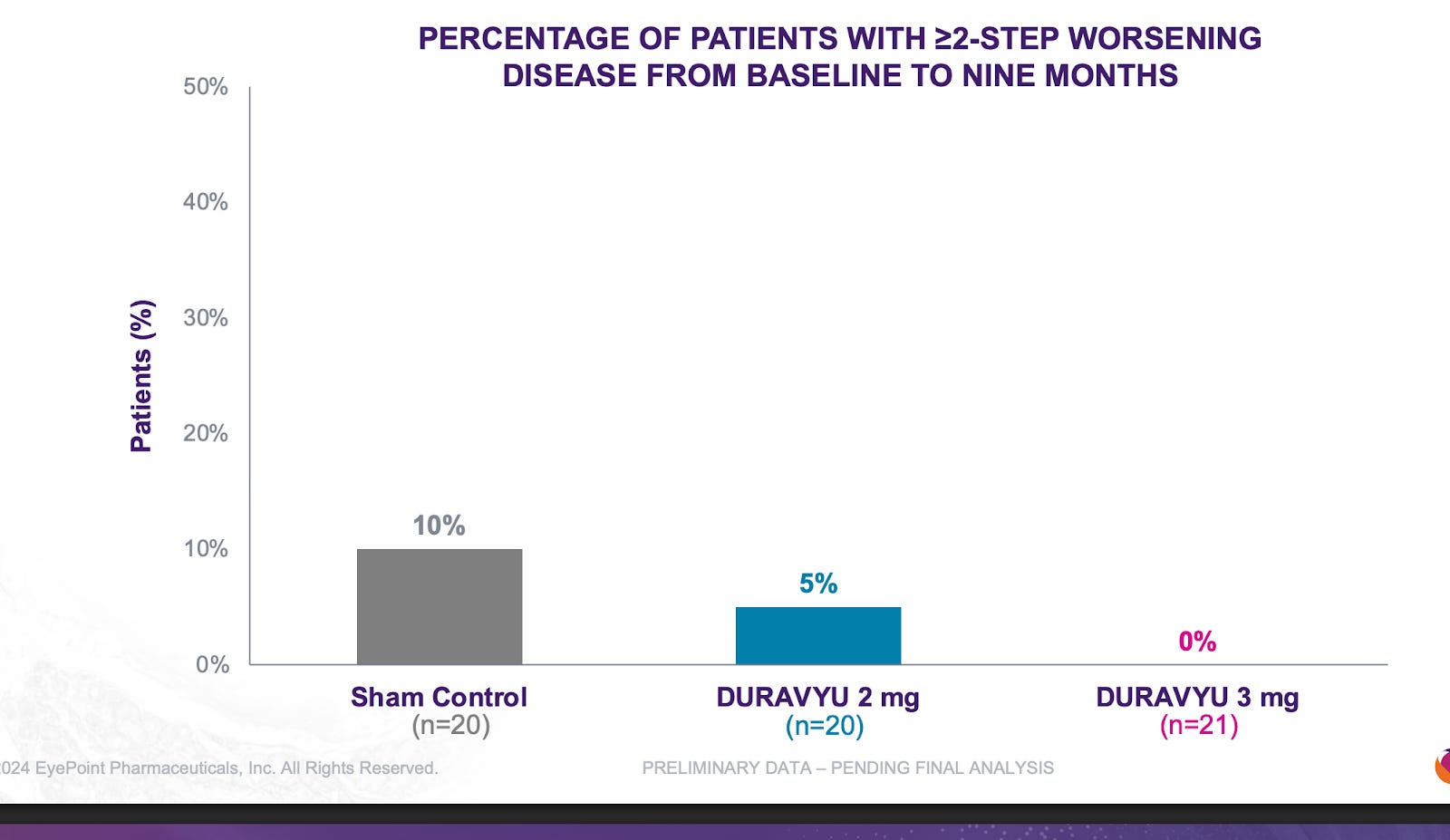
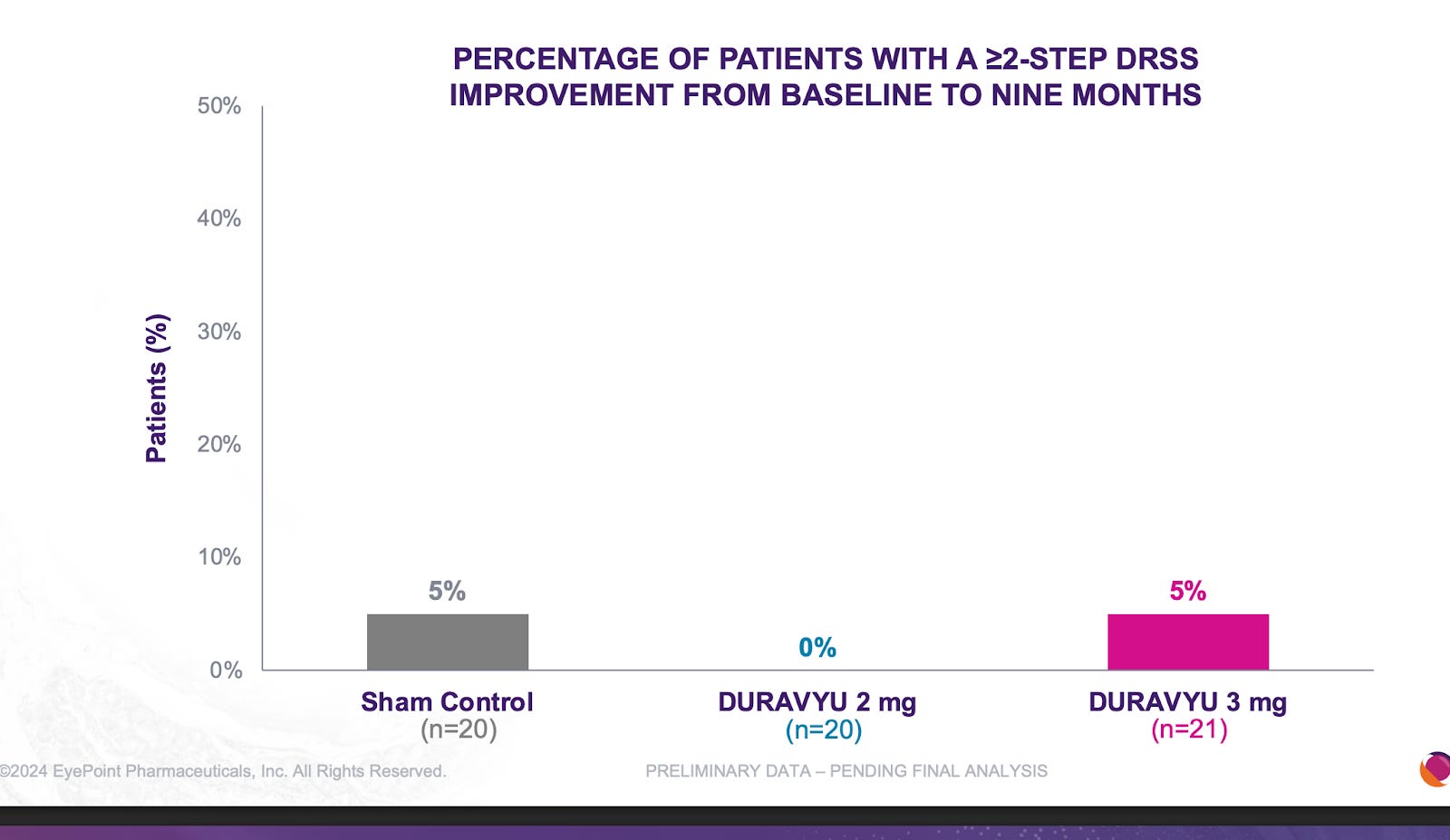
I’m not going to write them off. They have another phase 3 reading out mid year this year and likely will be positive based on the phase 2 results in Davio. But I worry about the injection frequency. 63% rescue freerate vs 70% in OCUL.
In my opinion,they can win. Beautiful you’re a doc/patient, and the injection frequency is -80% with one drug and -90% with another, there are few markets with “equivalent” drugs. Doctors have a preferred option. Furthermore, only one of the drugs will have a superiority label (again, matters for insurance) and data out to 12 months. So a Doc may feel more comfortable leaving a patient with OCUL. This is justified in the 2x market cap for OCUL.
**Market Case
If approved, the TKI will be the baseline therapy for many wAMD patients. A best in class TKI would be worth 5B+.
They’ll price at a premium to VEGF, likely at 2-3x Eylea to match their pricing
40% discontinuation rates over time ->they will expand this market
Commercialization into retina is a concentrated endeavor: Private equity takeover and strong relationships means a big pharma isn’t needed for wAMD imo.
From a TPP perspective, Axpaxli will supersede EYP-1901 by a margin, if both hit the phase 3. One less discussed strength of Axpaxli is the needle required for the procedure. EYPT’s is evidently larger thus introducing more pain and inheriting more risks from the procedure. Number of inserts is also an important consideration in which 1 insert/eye will minimize the vision distraction while streamline the repeated dosing (e.g., what if 2 inserts don’t synchronize well? re-dose or not?)
Comparison of TPP
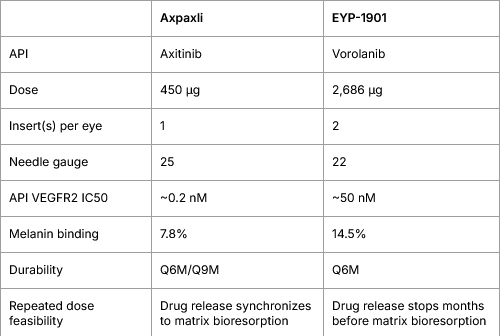
Comparison of Intravitreal Injection Needle Gauges
**Risks
KEY RISK 1: The new formulation
This Form IV has NEVER been tested in human before SOL-1/SOL-R. All the prior clinical studies (e.g., wAMD Australian phase 1, wAMD US phase 1, NPDR phase 1) used the old formulation. Bear in mind that there is always a chance where the inter-species translatability is off. If this ever happens, the efficacy in SOL-1/-R will be disappointing.
How big is this translational risk of the new formulation? Very small IMO. First, the FDA signed off the new formulation for the wAMD phase 3s in the SPA. OCUL must have submitted a bolus of preclinical evidence suggesting Form IV is superior to SAB-I, then FDA greenlighted. Second, the new NPDR HELIOS-3 trial also adopted the new formulation, after OCUL saw some blinded data from SOL-1 and some tidbits from SOL-R. This speaks to the confidence from both sponsor and regulator that this new formulation is highly likely to translate well between species.
There is also a minor chance of a severe AE damaging the program. Although we see no indication that Axpaxli should cause a major adverse event, people with wet AMD can have damaged eyes and even if Axpaxli itself doesn’t cause any damage, the injection itself can damage eyes quite terribly in rare circumstances.
KEY RISK 2: Control arm massively outperformed
There is also a possibility that the 2mg aflibercept arm may massively outperform expectations. In the real world many patients are able to extend their dosing out quite a lot, if the control arm exceeds expectations there’s a possibility that the price move on great Axpaxli data would be diluted. Two scenarios:
Delta becomes borderline statistically significant (say 63% vs 80%, delta = 15-20%). The stock will be punished moderately, because it opens the door for arguments that Eylea HD/Vabysmo would have outperformed Axpaxli in SOL-1.
A surprise miss of statistical significance brings the entire program into question.
Likelihood - Low. We have done our best inferring the control performance won’t be crazy but the clinical data & RWD are far from predictive due to the unique endpoint design and patient enrichment. The most convincing evidence that this is unlikely to happen is this quote from Pravin:

I guess the management won’t be satisfied with an overall 20% rescue, right?
This risk is likely the biggest one: Kaiser, the key OCUL KOL cited rescue rates much higher in the expectations. OCUL management cannot agree
“We’ve also looked at the IRIS database in the U. S. We’ve looked at the Aetna database in the U. K. And other databases in the U. K, for those of us, you know, now that we’re in the U. K. And people here will remember this. When Lucentis launched in the U. K, the reimbursement pathway, the reimbursed product pathway was that you presented as a naive patient, you got 3 monthly doses of Lucentis And then essentially, you were treated PRN thereafter. And you didn’t get reimbursed until you got 2,040 or worse, Which is exactly the patient population that we see in the comparator arm for ex Paxly. So we’re very, very confident That arm, that 30 to 50 percent of patients in that arm are going to proceed to a 15 letter loss”.
KEY RISK 3: Eylea NPDR Readthroughs
Ocular’s reported NPRD phase 1 data fell greatly short of Q8/12W Eylea. There isn’t a clear explanation for this phenomenon but it may be due to the greater total drying power than Eylea or the circulating VEGF in the eye.
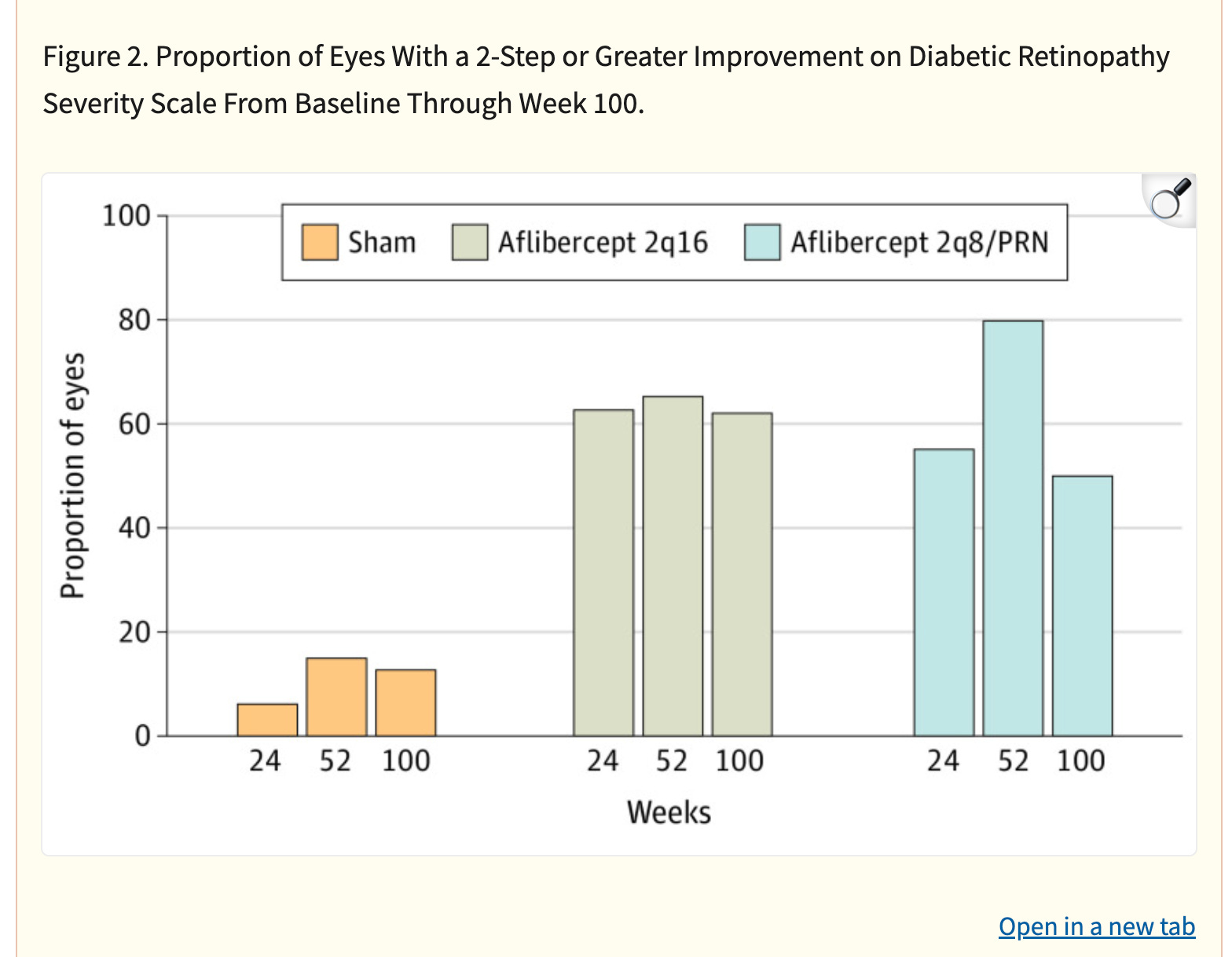
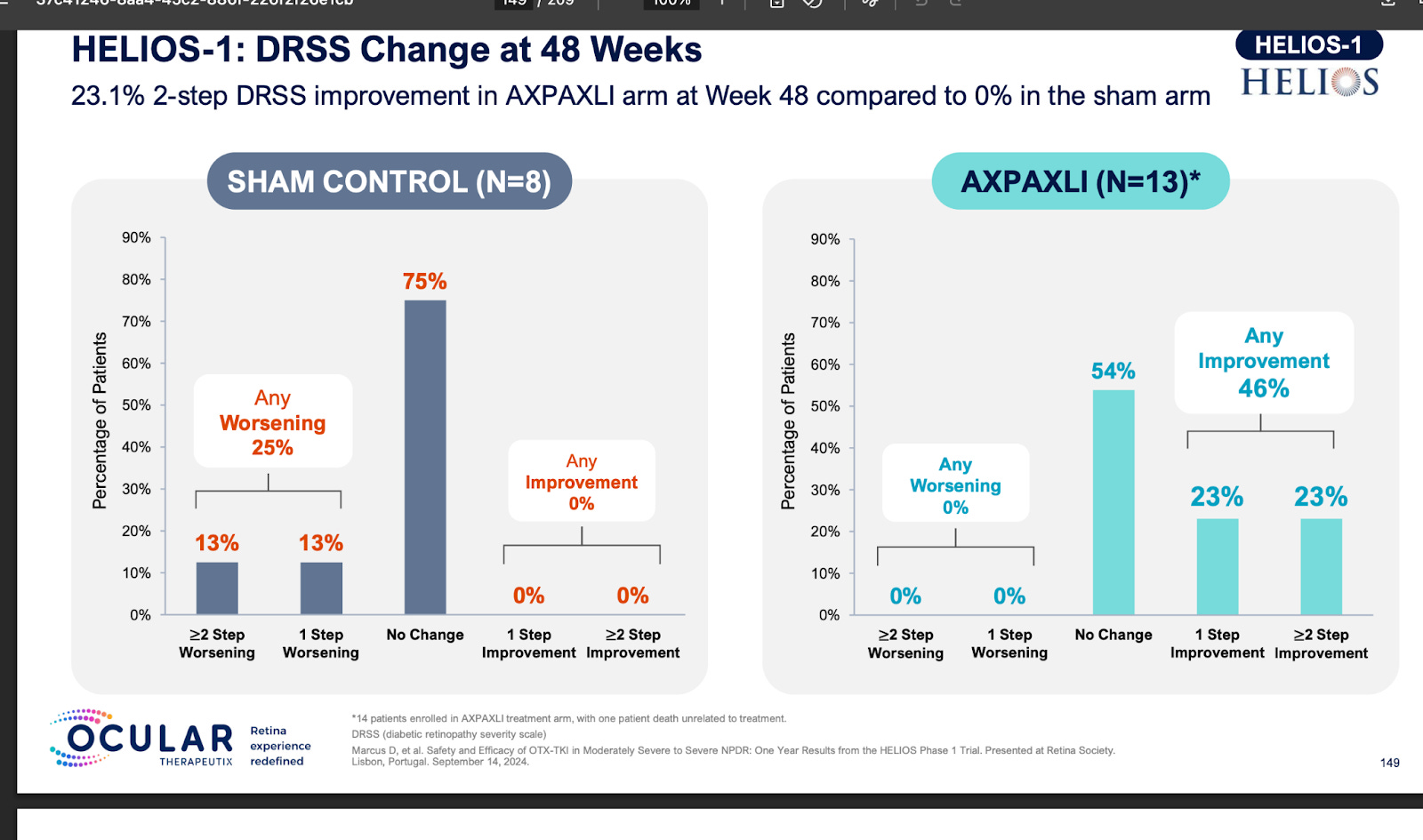
It is quite clear that TKIs < antibodies in NPDR. What is extremely interesting is that the Eyepoint NPDR data was far inferior to Oculars(though nothing like the Eylea Axpaxli gap). There seems to be no reason for this inferiority other than Duravu having worse long term VEGF suppression than Axpaxli, as both TKIs target the same important kinases for disease progression (If Duravu actually hit IL-6 you’d imagine it would beat Axpaxli in NPDR). It is of note that Ocular used the old SAB-1 formulation in their NPDR study.
Disclaimer:
The authors hold securities which benefit if the share price of Ocular rises. For entertainment purposes only. Not Financial Advice or whatever.
BalaBio: X
 | A guest post by
|